
Anyo Labs Molecular Generator
Importance and Limitations of Generative AI
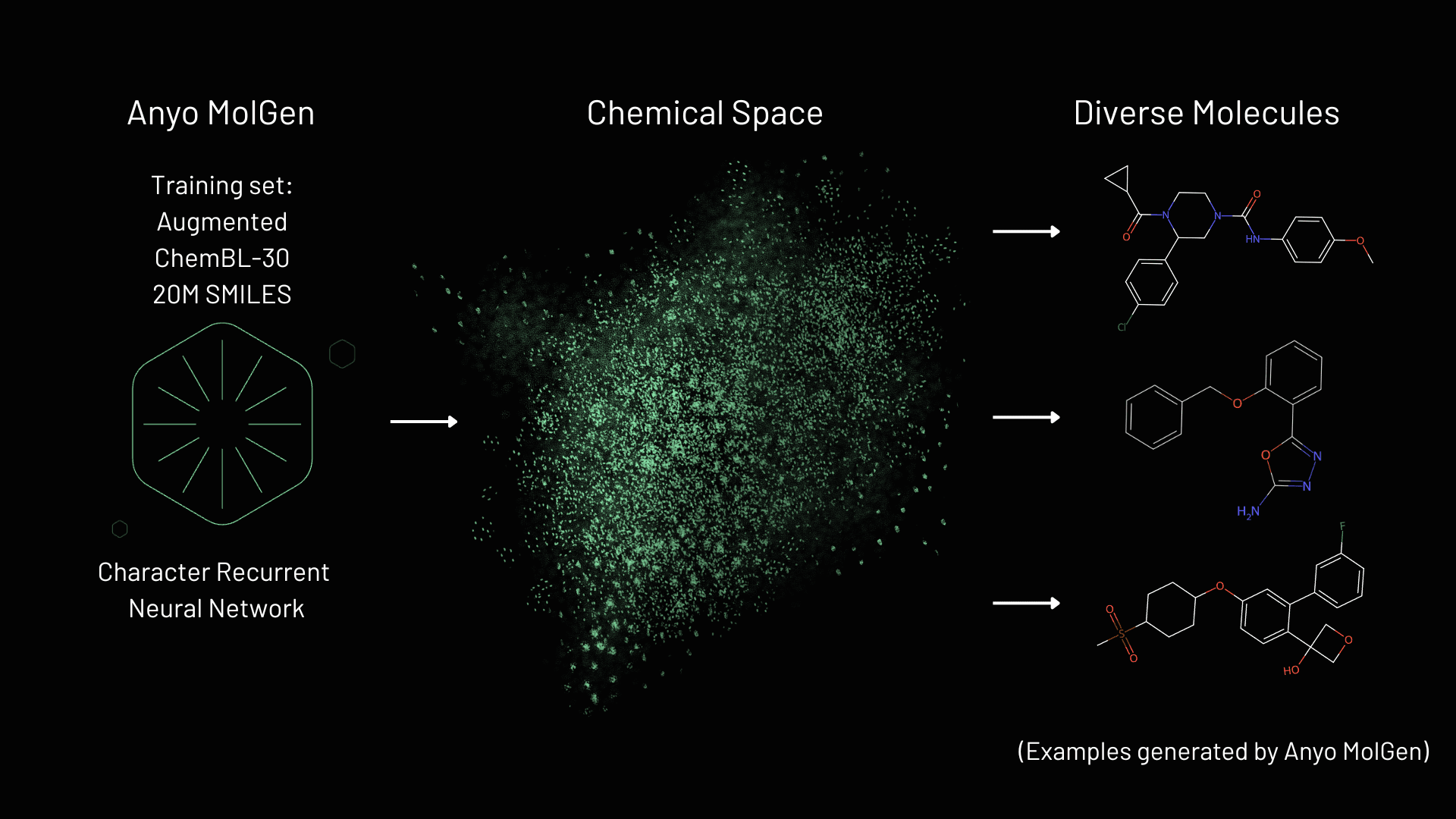
Anyo Labs molecular generator (MolGen) is an advanced generative AI engine designed to advance the early-stage drug discovery by generating novel, chemically valid, and synthesizable small molecules represented as SMILES strings. The tool leverages computational methods to create diverse molecular libraries from scratch, bypassing the limitations of existing static libraries limited in terms of size, and possibly diversity [1]. At the tools core is a character recurrent neural network (RNN), which was trained on an augmented set of 20 million bioactive molecules, enabling it to probe the vast chemical space of drug-like molecules. The potential space vastly exceeds traditional databases like GDB-17, which contains 166 billion molecules [2]. By generating tailored, diverse molecules, MolGen offers a dynamic alternative to screening before labor-intensive synthesis and testing, accelerating the discovery of lead candidates.
Generative AI in drug discovery
Generative AI (Gen AI) is critical for drug discovery, as it enables the rapid proposal of novel compounds to address unmet medical needs, potentially yielding treatments for complex diseases. Gen AI offers several key advantages over existing molecular libraries in drug discovery. Unlike traditional molecular libraries, which are limited to pre-existing compounds and often constrained by size and diversity, Gen AI can design novel molecules de novo, exploring vast chemical spaces to generate structures with tailored properties, such as improved binding affinity, selectivity, or drug-like characteristics. Gen AI models, trained on diverse chemical and biological data, can predict and optimize molecular properties, reducing reliance on time-consuming and costly high-throughput screening of physical libraries [3]. Additionally, Gen AI enables rapid iteration and customization, allowing researchers to generate molecules that target specific protein binding sites or address unmet therapeutic needs, often producing innovative scaffolds that may not exist in conventional libraries. This flexibility and ability to explore uncharted chemical space make Gen AI a powerful tool for accelerating hit identification and lead optimization compared to the static nature of existing molecular libraries.
However, existing generative AI models face significant limitations. Most, such as those based on variational autoencoders (VAEs) or generative adversarial networks (GANs), generate libraries with limitations in structural diversity [4,5]. For instance, VAE models benchmarked in MOSES (trained on ZINC’s ~220,000 molecules) tend to overfit their training distributions, producing redundant or similar structures [4,6]. Additionally, slow generation speeds hinder efficient exploration of chemical space, constraining the discovery of unique lead candidates [4,7]. These limitations underscore the need for scalable and efficient generative AI models capable of producing larger and more diverse molecular libraries.
Anyo Labs drug discovery pipeline
The combination of Anyo MolGen with the ultra-fast Scoring method [8], encompasses a proprietary method that enhances computational efficiency and scalability in molecular generation:
- High Validity: Anyo MolGen generates 95.4% valid molecules, ensuring high-quality outputs (measure on 1 million samples).
- High Uniqueness: Over 98.9% of valid compounds are unique, minimizing redundancy across large-scale generation.
- High Novelty: Our benchmarking indicated that over 96.5% of the generated compounds were not in the training set.
- Excellent Synthesizability: Despite sampling novel compounds, the high synesthetic accessibility of the generated molecules was acknowledged by our experimental expert partners.
- Exceptional Speed: Anyo MolGen generates >5,700 compounds per second on a single NVIDIA A100 node, significantly outpacing many competing models [4,7].
The Scoring method, detailed in our 2025 study [8], optimizes for both accuracy and scalability, in combination with Anyo MolGen allows the user to explore unseen parts of the chemical space. A seed-free approach and high throughput enable it to explore vast, diverse chemical spaces, making it ideal for early-stage drug discovery. This efficiency sets Anyo MolGen apart from competitors limited by smaller library sizes and diversity, positioning it as a highly scalable solution for navigating the vast chemical space.
Real-World Validation: Internal Project Success
The capability of out drug discovery pipeline was validated in an internal hit identification project where hundreds of millions of novel compounds were designed, 200 compounds were selected based on their inhibitory potential and their synthetic possibility evaluation was conducted by organic chemists. Remarkably, all 200 were deemed synthesizable at the first glance, with 150 classified as unproblematic for synthesis. Anyo then synthesized 32 novel compounds of which all were successful and delivered for experimental testing. This high success rate for synthetic feasibility demonstrates the high capability of MolGen model for producing practical, high-quality molecules suitable for laboratory development. The project highlights a key advantage: MolGen’s vast, diverse output allows researchers to apply straightforward filters for synthetic feasibility, streamlining the transition from virtual molecules to lab-ready candidates. Compared to models in the MOSES benchmark, which often yield lower validity and diversity [4], Anyo MolGen’s high uniqueness (>%) and validity ensure a greater proportion of synthesizable hits, accelerating the drug discovery pipeline.
Applications in Drug Discovery
Applying a generative AI to Anyo Labs toolkit supports multiple stages of drug discovery:
- Novel Lead-Like Hit Generation: As shown in the internal project, MolGen produces diverse, synthesizable compounds, providing a rich pool for early-stage screening.
- Hit Identification via Similarity Search: Generated “hits” can be cross-referenced to immediately purchasable datasets from different vendors like MolPort, Enamine etc. to identify molecules with similar properties, while being easily synthesized.
- Hit-to-Lead and Lead Optimization: By providing the model with an active scaffold, Anyo MolGen becomes an analogue generation tool. Applying downstream filtering a pipeline that optimizes molecules for multiple pharmacokinetic properties can be created, refining leads for clinical development.
These applications underscore Anyo's generative AI's role as a comprehensive tool for advancing drug discovery from hit identification to lead optimization.
Looking Ahead:
Article titled “Exploring Chemical Space A forthcoming”, set to be published later this month, will provide a detailed analysis of Anyo MolGen’s ability to explore chemical space, estimated at a minimum of hundred Trillion unique molecules, with theoretical upper limits surpassing size of the drug-like chemical space. This study will quantify the tools diversity, reporting Tanimoto diversity scores up to 0.89 for SMILES and scaffolds, and introduce innovative methods, such as ecological species richness estimators (e.g., Chao1, ACE), to estimate its vast generative capacity.
References
[1] Manan, A., Baek, E., Ilyas, S., & Lee, D. (2025). Digital Alchemy: The Rise of Machine and Deep Learning in Small-Molecule Drug Discovery. International Journal of Molecular Sciences, 26(14), 6807. https://doi.org/10.3390/ijms26146807
[2] Ruddigkeit, L., et al. (2012). Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. Journal of Chemical Information and Modeling, 52(11), 2864–2875. https://doi.org/10.1021/ci300415d
[3] Walters, W. P., & Murcko, M. (2020). Assessing the impact of generative AI on medicinal chemistry. Nature Biotechnology, 38(2), 143–145. https://doi.org/10.1038/s41587-020-0418-2
[4] Polykovskiy, D., et al. (2020). Molecular sets (MOSES): A benchmarking platform for molecular generation models. Frontiers in Pharmacology, 11, 565644. https://doi.org/10.3389/fphar.2020.565644
[5] Gómez-Bombarelli, R., et al. (2018). Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Science, 4(2), 268–276. https://doi.org/10.1021/acscentsci.7b00572
[6] Jin, W., et al. (2018). Junction tree variational autoencoder for molecular graph generation. Proceedings of the 35th International Conference on Machine Learning (PMLR), 80, 2323–2332. http://proceedings.mlr.press/v80/jin18a.html
[7] Segler, M. H. S., et al. (2018). Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Central Science, 4(1), 120–131. https://doi.org/10.1021/acscentsci.7b00512
[8] Mahdizadeh, S. J., & Eriksson, L. A. (2025). iScore: A ML-based scoring function for de novo drug discovery. Journal of Chemical Information and Modeling, 64(7), 2560–2571. https://doi.org/10.1021/acs.jcim.4c02192